You must be signed in to read the rest of this article.
Registration on CDEWorld is free. You may also login to CDEWorld with your DentalAegis.com account.
With each day, advances in surgical and implant technology enable dentists to treat an esthetically demanding patient population. More and more patients are seeking surgical care to meet their esthetic expectations. Surgeons need to be mindful of the surgical aspects of care and also need to be aware of systemic and local factors that may impede the delivery of that care.
Surgeons may be faced with providing periodontal plastic or implant therapy to patients with gingival manifestation of systemic conditions. These conditions often present with features of desquamative gingivitis (DG), which was once believed to represent a disease entity. However, today, DG is used to describe clinical features of various local or systemic diseases or disorders that result in chronic gingival lesions characterized by epithelial desquamation, erythema, ulceration, and/or vesiculobullous lesions of the gingiva. Often, other oral tissues also are involved.1 Mucocutaneous disorders include such disease entities as lichen planus, graft-versus-host disease (GVHD), pemphigoid, pemphigus vulgaris (PV), lupus erythematosus (LE), erythema multiforme, and linear IgA disease (LAD) (Table 1). Surgeons should be able to recognize these disorders and possess the tools necessary to treat these disorders so that they can render the appropriate surgical care.
Desquamative Gingivitis (DG)
The etiology of DG may range from mechanical trauma, such as deliberate factitious injury or inadvertent toothbrush trauma, to bacterial, viral, or fungal infections, or to reactions to various allergens, such as toothpaste, mouthrinse, chewing gum, and candy. In addition, a number of systemic diseases have been associated with chronic erythematous or erosive gingival lesions. Consequently, DG does not represent a specific disease entity; rather, it is a clinical feature of many oral and systemic diseases and disorders. Some systemic diseases, such as psoriasis, or certain chronic granulomatous diseases, such as sarcoidosis, Crohn’s disease, Wegener’s granulomatosis, and plasmacytosis, may manifest with clinical features very suggestive of DG although actual desquamation may not be present. For the most part, however, various vesiculobullous mucocutaneous disorders often are found to be associated with DG.1
Approximately 80% of the time, DG represents a clinical characteristic of mucous membrane pemphigoid (MMP) or oral lichen planus (OLP).1,2 However, a significant number of other dermatologic disorders sometimes affect the gingival and oral mucous membranes. These include, but are not limited to, PV, LAD, chronic ulcerative stomatitis (CUS), GVHD, frequent or recurrent erythema multiforme, and LE. Diagnosis of these conditions is based on medical and dental history, clinical examination, and, often, biopsy to include both histopathologic and immunofluorescent evaluations.3 This article will limit the discussion to the mucocutaneous diseases mentioned above.
The Diseases
Lichen Planus
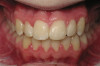
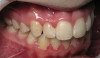
Lichen planus is the mucocutaneous disorder seen most often in the oral cavity (Figure 1). It is a condition of unknown etiology, although a strong autoimmune component plays a role in lesion formation. The disorder usually develops between the ages of 40 and 70 years, and women are affected more often than men.4 Chainani-Wu et al5 reported a male-to-female ratio of 1:2 in 209 patients, while data from the Stomatology Center at Baylor College of Dentistry suggest a 3:1 female-to-male ratio in greater than 800 patients.6
More than 100 years ago, lichen planus was recognized as a skin disease; however, an awareness of frequent oral involvement developed later. Studies suggest that more than one third of individuals with lichen planus of the skin will also present with oral lesions, while those with oral lesions will present with skin lesions approximately 12% to 14% of the time.7
Lichen planus can occur on any mucocutaneous tissue, and a vulvo-vaginal-gingival lichen planus syndrome has been reported in 20% to 57% of women with gingival lesions. A peno-gingival syndrome also exists, although this appears to occur in approximately 5% of men with OLP.7-10
Skin lesions have been described as purple, pruritic, polygonal papules (known as the “4Ps”) (Figure 2) that often occur on the flexor surfaces of the body. Oral lesions were classified by Andreason in six different forms: reticular, papular, plaque-like, atrophic, ulcerative, and bullous.11Lesions that were atrophic, ulcerative, or bullous were considered by others to be erosive because these lesions were usually painful and tended to slough or to form vesiculobullous lesions, which later slough, leaving ulcerations.
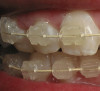
Today, however, there is a trend toward consolidating the types of OLP into the reticular (reticular, papular, plaque-like), erythematous (atrophic), and erosive (ulcerative, bullous) forms7 (Figure 3 and Table 2). Some have limited the forms to reticular (reticular, plaque-like) and erosive (atrophic, bullous, and ulcerative).12 Variations in terminology make it important to define terms when discussing the types of OLP with other healthcare providers regarding shared patients.
OLP can occur on any mucosal surfaces of the oral cavity; however, the buccal mucosa is the most common site. The tongue and lips also may be involved with some frequency. Gingival involvement is relatively common, and Stomatology Center data suggest more than 50% of patients with OLP referred to a periodontal practice may have gingival lesions with nearly 20% of lesions confined entirely to the gingiva and manifesting as DG.6
Diagnosis is based on previous history, clinical appearance, histologic examination, and direct immunofluorescence studies. Histopathologic features of OLP include hyperkeratosis, liquefaction degeneration of the basal cell layer of epithelium, saw-toothed or shortened rete ridges, and an intense band-like inflammatory infiltrate of predominate lymphocytes in the lamina propria with or without colloid bodies7 (Figure 4).
Many conditions, including dysplastic precancerous lesions, may present with some of these characteristic histologic features. Consequently, it has been suggested that the definitive diagnosis of lichen planus be reserved for lesions with virtually all of the clinical and histopathologic features, while lesions with some but not all features are alluded to as lichenoid lesions or lichenoid mucositis.13 Using these criteria, lichenoid lesions were associated with transition into squamous cell carcinoma (SCC), while OLP lesions were not.13 However, it should be noted that the criteria specified by this method do not necessarily represent the features of OLP seen in clinical practice.
Immunofluorescence studies are not diagnostic, but they may be very beneficial in confirming a diagnosis of OLP. In skin biopsy samples, lichen planus lesions often present with positive cytoid bodies in the lamina propria. Although these may be found in OLP as well, the more predominant feature is a linear pattern of antifibrin or antifibrinogen in the basal membrane zone.4
Although this is somewhat controversial, OLP has been associated with other systemic maladies, such as stress, chronic active hepatitis (especially if hepatitis C induced), and GVHD.4,14-16,17 However, considerable evidence suggests that any relationship with these diseases may represent a lichenoid drug reaction to medications being used in the treatment.18
Case reports have ascribed lichenoid reactions to more than 50 drugs, and it is likely that any drug can induce this phenomenon on rare occasions. Nonsteriodal anti-inflammatory drugs (NSAIDs) and medications used in management of hypertension and cardiovascular disease are associated most frequently with induction of a lichenoid drug reaction.19
Contact lichenoid reactions have been traced to various metallic and nonmetallic dental restorative materials with allergy to mercury, nickel, and gold being most common. Contact reactions also have been associated with various flavoring agents and preservatives used in dental hygiene products and foods, with the flavoring agent cinnamic aldehyde most frequently identified as the causative agent.20
Considerable controversy exists in regard to the possible relationship between OLP and SCC. Numerous case reports have described such a relationship, although closer evaluation of histologic specimens suggests that premalignant epithelial dysplasia may present with some, but not all, features of OLP.13 Others have described the development of SCC in lesions previously diagnosed as OLP.21 Although this issue has not been resolved, clinicians must be aware that patients who receive a diagnosis clinically, histologically, or by direct immunofluorescence, as well as patients with lichenoid mucositis, may be at a slightly increased risk for subsequent SCC.5,17
Graft-Versus-Host Disease (GVHD)
Hematopoietic cell transplantation has markedly improved the prognosis for patients who have hematologic malignancies, hematologic immune states, and bone marrow failure. This therapy is not without risk, however, and GVHD is a frequent adverse effect of allogeneic hematopoietic stem cell or bone marrow transplantation (Figure 5). The condition occurs when the engrafted cells recognize host cells as “foreign” and attempt to reject the host cells. This rejection process may begin within days of hematopoietic transplantation, or its occurrence may be delayed.
Traditionally, GVHD that occurs within the first 100 days after engraftment is considered to represent acute GVDH, while chronic GVHD arises 100 days or more after engraftment.4 There are characteristic differences in the two processes, however, and chronic GVHD offers the worse prognosis. Disease may progress from acute to chronic, or chronic GVHD may arise de novo or after a period of quiescence.22 GVHD may occur in 20% to 90% of hematologic graft recipients depending on the degree of histocompatibility match between the donor and recipient. The primary target organs are the skin, liver, and gastrointestinal tract. The oral cavity may be involved in 35% to 90% of affected individuals.
Diagnosis of oral GVHD is based on the presence of erosive mucocutaneous lesions, with or without biopsy confirmation, and abnormal liver function tests. Oral signs and symptoms may include features that are indistinguishable from LE, systemic sclerosis, Sjögren’s syndrome, or erosive lichen planus. The buccal and labial mucosa and tongue are affected most often. Approximately 50% of individuals will have gingival lesions consistent with DG; however, gingival ulceration is rare.23
Chronic GVHD profoundly affects morbidity and quality of life in patients who have this condition long term. Multiple organ systems are likely to be involved, and systemic therapy is probable. Patients may receive immunosuppressant drugs, such as cyclosporine, tacrolimus, sirolimus, corticosteroids, and azathioprine, for months, years, or even life.24 Perhaps in part because of these medications, patients receiving hematopoietic transplant have a two- to eightfold greater chance of developing secondary hematologic malignancies than the general population does.24 Solid organ malignancies, including oral SCC, also increase in frequency, and susceptibility appears to increase with time.25
Chronic Ulcerative Stomatitis (CUS)
CUS is a newly described idiopathic condition that presents in the oral cavity and sometimes the skin, with ulcerations and inflammatory features highly suggestive of erosive or erythematous lichen planus. To date, approximately 40 cases have been reported in the medical and dental literature, although the true incidence is probably greater. Oral lesions occur on the tongue, buccal mucosa, and gingiva, with gingival lesions usually manifesting as DG.26,27 The average age of those with the condition is 60 years, and women appear to be affected more often than men.28
Histologically, biopsy specimens present with features suggestive of OLP or lichenoid mucositis, and direct immunofluorescence is essential to establish the diagnosis, revealing intranuclear speckled staining by immunoglobulin G (IgG) antibodies in the cells of the basal and lower spinous epithelium.29 Indirect immunofluorescence may reveal circulating stratified epithelium-specific antinuclear antibodies, although recent evidence indicates that these antibodies occasionally may be found in various types of lichen planus.30,31 Normal oral tissue also may show positive staining. Several case reports suggest CUS is less responsive to local therapy using topical corticosteroids or systemic corticosteroids, and hydroxychloroquine sulfate therapy may be required to achieve remission. Lesions may recur when therapy is discontinued.26,32
Mucous Membrane Pemphigoid (MMP)
MMP is a heterogenous group of blistering autoimmune disorders of unknown etiology. Several subsets have been identified, with one believed to cause oral lesions exclusively while others may involve other mucous membranes and skin.33,34
MMP usually occurs in individuals older than 50 years, although it has been reported in children and young adults.35-37 It is more common in women than men at a ratio of 2:1.36 Any mucosal tissues can be involved; however, oral and ocular lesions are far more frequent than lesions at other sites.
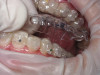
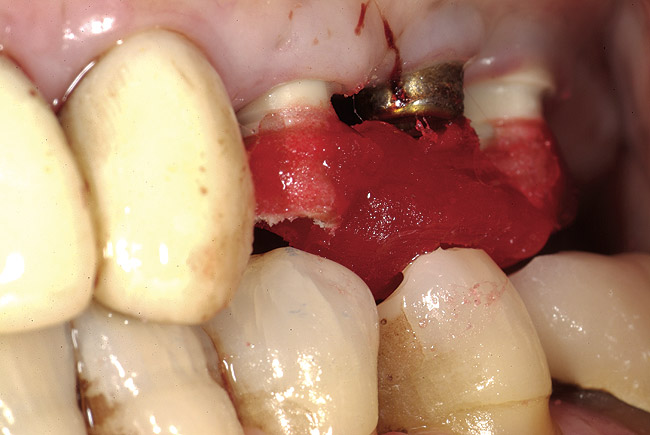
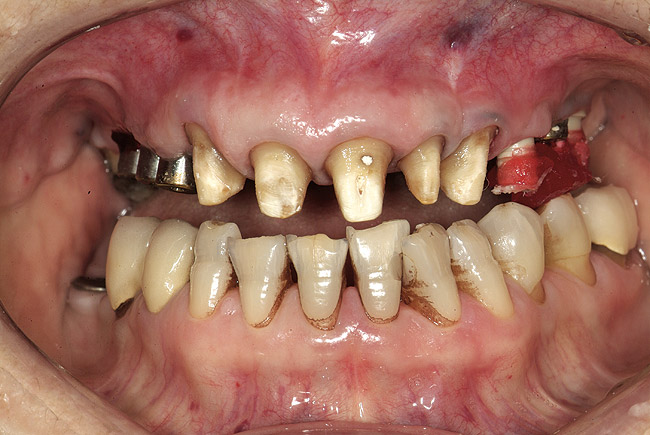
Oral manifestations feature formation of vesiculobullous lesions, a positive Nikolsky’s sign, and DG (Figure 6). Nearly all individuals who develop MMP will have DG, and the gingiva may be the only site of involvement in more than 50% of cases38 (Figure 7).
Rarely, a few drugs (captopril, carbamazepine, clonidine, furosemide, penicillamine, practolol) have been reported to cause a pemphigoid-like reaction. However, these reactions are found most frequently on skin, mimicking bullous pemphigoid, a similar disease with predominant skin manifestations.36
Patients with MMP may present with eye lesions, such as entropion, trichiasis, ulceration, and symblepharon formation. Blindness also may occur (Figure 8).38 Consequently, an important part of management is referral of patients who have recently received a diagnosis of oral MMP to an ophthalmologist for examination and appropriate treatment, if necessary. MMP diagnosis is based on clinical appearance, histopathologic examination, direct immunofluorescence, and, sometimes, indirect immunofluorescence.38 The dominant histopathologic feature is subbasilar separation of epithelium from underlying connective tissue, with a relatively mild mixed inflammatory cell infiltrate in the lamina propria (Figure 9).38 This feature also may be detected in similar blistering diseases, such as epidermolysis bullosa, epidermolysis bullosa acquisita, and LAD. Consequently, direct immunofluorescence is important in helping confirm the diagnosis. Direct immunofluorescence findings include a linear pattern of basement membrane zone with anti-C3 and IgG33,39 (Figure 10). However, because other diseases also may present with these direct immunofluorescence features, more sophisticated studies using direct immunofluorescence on salt-split skin or computer-aided fluorescence overlay antigen mapping with laser scanning confocal microscopy should be used to enhance the ability of the immunopathologist to differentiate between subtypes of MMP and the other related diseases.40-42 These sophisticated diagnostic tools may become increasingly important as evidence surfaces indicating that certain subsets of MMP may be associated with an increased risk of malignancies.43
Indirect immunofluorescence may be of benefit in identifying circulating autoantibodies. However, not all patients with MMP have identifiable circulating antibodies, and positive findings are present in only 60% to 65% of their blood samples.43
Linear IgA Disease (LAD)
LAD is a chronic, usually acquired, autoimmune subepithelial blistering disease of skin and mucous membranes. On occasion, lesions are confined to the oral cavity or precede cutaneous lesions. Erosive lesions of the tongue, buccal mucosa, and palate are common, and gingival lesions may manifest as DG.29,44-46
LAD is characterized by a linear pattern of IgA autoantibody deposition in the basement membrane zone.47 Skin lesions manifest as urticarial plaques, vesicles, and bullae, and oral lesions are often vesiculoulcerative, with or without DG.48
LAD is considered by some to represent a variant of pemphigoid.49 However, many authorities believe the condition is a unique, acquired disease usually induced by an adverse reaction to medications, such as vancomycin, amiodarone, lithium, or NSAIDs. Others have described the development of LAD in association with inflammatory bowel disease, other autoimmune diseases, and malignancy.50-52 Often, however, the trigger is unknown.53
The condition occurs in children rarely (linear IgA bullous disease of childhood) and more often in adults (LAD).50 Clinically, lesions mimic MMP or dermatitis herpetiformis. Histopathologic features are similar to MMP, although papillary microabscesses and a predominant neutrophilic inflammatory infiltrate may be found.
Definitive diagnosis usually is made by direct immunofluorescence analysis, revealing a homogenous linear pattern of IgA deposition in the basement membrane zone. Occasionally, direct immunofluorescence also will be weakly positive for IgG and/or IgM.
Clinical reports indicate LAD is more treatment-resistant than MMP, and potent systemic therapy may be necessary to control the condition.49
Pemphigus
Pemphigus is a family of autoimmune diseases that affect skin and mucous membranes. It occurs in various forms, with the mildest presenting as pemphigus erythematosis, which is characterized by mild generalized cutaneous erythema. Pemphigus foliaceous is also mild, although some vesicle and bullae formations may occur. The condition rarely involves the oral cavity. One pemphigus foliaceous variant, fogo selvagem, tends to occur in epidemics, and a viral etiology is suspected.54
Paraneoplastic pemphigus (PNP) affects the oral cavity extensively, while extraoral lesions may be present. In the mouth, it is characterized by formation of nonspecific vesiculobullous lesions that may mimic other mucocutaneous diseases, such as OLP, LE, erythema multiforme, toxic epidermal necrolysis, and PV.55,56
Lesion formation is triggered by the presence of an underlying malignant or a benign tumor in the body and often may represent the first warning that a tumor is present. PNP has been widely reported secondary to the presence of non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, thyoma, sarcoma, IgM myeloma, and Castleman’s disease (a benign but progressive lymphoproliferative disorder). Diagnosis of PNP is sometimes difficult, and evidence suggests it is best identified using indirect immunofluorescence on nonstratified epithelium, such as rat bladder, which is rich in desmoplakins.56 Oral lesions are resistant to successful treatment unless the underlying tumor can be eliminated.
Pemphigus Vulgaris (PV)
PV is the most aggressive, potentially lethal form of pemphigus. It is an autoimmune disorder that is most prevalent between the fourth and sixth decades of life, although the condition has been identified in children. Both men and women may develop it, and a genetic predisposition is indicated by its increased incidence in Ashkenazi Jews and individuals of Mediterranean origin.57
Cutaneous involvement can be severe, and mortality rate is relatively high despite development of more effective treatment modalities. Bullous skin lesions break quickly, often leaving generalized erosions that may lead to septicemia, fluid loss, and electrolyte imbalance. Oropharyngeal lesions and involvement of the genitalia and eyes can be debilitating.58
The oral cavity often is affected early in the disease process and, on occasion, may be the only site of involvement.59 Lesions most often are seen on the buccal mucosa, palate, tongue, and lips. They manifest as quickly bursting blisters, leading to erosions and ulcer formation (Figure 11). Gingival lesions may appear first as isolated blisters or areas of tissue sloughing, but, as the condition progresses, severe DG may occur60 (Figure 12). Nikolsky’s sign is prominent, and lesions sometimes may remain confined to the gingiva61 (Figure 13).
Diagnosis is based on clinical features, histopathologic evaluation, and direct and indirect immunofluorescence. Histologic features include characteristic intraepithelial ballooned Tzanck cells and acantholysis, causing a suprabasilar epithelial separation (Figure 14). Direct immunofluorescence reveals antibodies to complement various immunoglobulins, most commonly IgG.57 This reaction is found intracellularly in epithelium, creating a distinctive direct immunofluorescence appearance, which is sometimes referred to as the chicken wire effect (Figure 15). Indirect immunofluorescence can help determine the severity of the antigen-antibody reaction and monitor treatment progress.57
PV is representative of an autoimmune attack against desmosomes and hemidesmosomes that are responsible for epithelial cell adhesion. Desmogleins are the desmosomal proteins that are reactive to autoimmune antibodies in PV. The presence of circulating antibodies involving desmogleins can be detected using enzyme-linked immunosorbent assay (ELISA) techniques.62
Three types of desmoglein are found in stratified squamous epithelium (types 1, 2, and 3). In skin and mucosa, desmoglein type 1 is expressed in suprabasal cell layers, desmoglein type 2 is found in the basal layer only, and desmoglein type 3 appears in the basal and immediate suprabasal layer.62 In oral mucosa, desmoglein type 1 is expressed minimally. Consequently when only desmoplakin type 3 antigens are identified, lesions may be found exclusively in the oral cavity. If antibodies to both desmoglein types 1 and 3 are elevated, skin lesions are likely. A pair of recent case reports documents epitope switching from oral PV with desmoglein type 3 antibodies to mucocutaneous disease, showing the presence of antibodies to both desmoglein types 1 and 3.61,63
Several drugs, including amoxicillin, penicillin, cephalosporins, quinolones, captopril, and penicillamine, have been reported to occasionally precipitate a pemphigus-like adverse reaction, and a relatively strong relationship has been identified between onset of PV and presence of other autoimmune diseases (bullous pemphigoid, lichen planus, scleroderma, rheumatoid arthritis, systemic LE, and others).57,64
Lupus Erythematosus (LE)
LE is a heterogeneous connective tissue disease. LE has been described as the ultimate autoimmune disease, with the systemic form featuring systemic circulating antinuclear antibodies against native DNA, as well as antigens, such as extractable nuclear, Smith, SSA, and SSB. Antigen-antibody complexes are deposited in tissues either locally or systemically.
Although the pathogenesis is not yet fully understood, LE is believed to represent the interaction of genetic, environmental, and hormonal factors.65 The condition may involve multiple body systems, and it presents with various manifestations. Discoid lupus erythematosus (DLE) is a condition of the skin and oral mucosa characterized by the presence of raised mixed white and erythematous plaques; it may represent the earliest disease stage, although DLE does not always precede the other forms.
Cutaneous LE may be classified as chronic, subacute, or acute. Chronic cutaneous LE may be localized or generalized. Discoid skin lesions are present, and the oral cavity may be involved. Oral lesions manifest as irregular white keratotic plaques or as lesions with an erythematous or ulcerated center surrounded by radiating white striations (Figure 16). They may occur on any oral mucosal surface.66 The gingiva may be involved, creating the appearance of DG, although desquamation is not always present (Figure 17).67 On buccal mucosa, the mixed red and white lesions may mimic OLP clinically and histologically, as well as on direct immunofluorescence. Lesions may occur exclusively in the oral cavity, or the oral cavity may be the initial site of lesions. More commonly, they are found in conjunction with cutaneous manifestations.68
Subacute cutaneous lupus erythematosus (SCLE) may manifest as annular discoid type plaques, as papulosquamous plaques, or as vesiculobullous lesions. The lesions are more likely to be generalized, and mild systemic symptoms may be present. The condition may not change for years or may evolve into systemic lupus erythematosus (SLE).
Acute cutaneous lupus (ACLE) features the classic malar rash, an erythematous patchy or plaque-like lesion usually bilateral and crossing the bridge of the nose. Patients often have generalized cutaneous involvement and are likely to meet the diagnostic criteria for SLE.69
SLE is capable of causing extensive damage to any bodily organ. Oral LE lesions may appear both clinically and histologically similar to OLP, and a lupus-lichen planus overlap syndrome has been reported. Oral ulcerative lesions may be more common in SLE than the other LE forms.69 Lesions may be found on the hard and soft palates, buccal mucosa, gingiva, and vermillion border of the lips. They are often painless; however, some individuals describe chronic burning and oral soreness.69
Outbreak of lupus lesions may be triggered by infections, elevated temperature, concurrent systemic diseases, and exposure to ultraviolet light or drugs. Many drugs have been indicted as precipitating an outbreak; however, the most common include procainamide, hydralazine, sulfonamides, antibiotics (penicillin and tetracycline), antimalarial agents, Dapsone, gold, and griseofulvin (Table 3). Ironically, the latter four drugs and medication groups often are used to treat other mucocutaneous diseases.70
SLE and DLE are more common in women, especially those with African descent, often in the third decade of life.71 SLE is associated with various clinical presentations, including weight loss, arthralgia, malar (butterfly) rash, nephrotoxicity, lupoid hepatitis, autoimmune thrombocytopenia, leukopenia, and aplastic anemia. Oral ulcerations are found in more than 40% of individuals with SLE, and, on occasion, oral discoid lesions may represent the earliest manifestation of SLE.72,73
Histologic features include hyperkeratosis with keratin plugging, atrophy of rete ridges, liquefaction degeneration of the basal epithelial cells, and thickening of the basement membrane. The lamina propria presents with a band-like lymphocytic inflammatory infiltrate separated from the basement membrane zone by a patchy PAS positive cell-free zone. Direct immunofluorescence may reveal granular deposits of multiple immunoglobulins, complement and fibrinogen in a subbasilar linear band (lupus band), and this band may be found in uninvolved tissue. Indirect immunofluorescence may reveal the presence of anti-DNA, anti-RNA, and antinuclear antibodies.66,72
Erythema Multiforme
Erythema multiforme is an acute mucocutaneous vesiculobullous inflammatory disease with distinctive cutaneous lesions (target lesions). Mucosal lesions can occur, involving the eye, oral cavity, and genitalia. The condition is usually self-limiting, although persistent and multiple recurrent forms have been described.
The etiology is unknown, although exposure to several drugs including antibiotics and a recent episode of herpes simplex virus (HSV) infection, mycoplasma pneumonia, or occasionally other bacterial or viral infections have been described as triggers. The presence of underlying systemic diseases and/or disorders also can generate an erythema multiforme outbreak.
HSV outbreak 2 to 3 weeks before the outbreak of initial lesions has been reported in 35% to 65% of cases.74,75 As a result of reports of this nature, several studies have described control of recurrent erythema multiforme by long-term use of acyclovir or other antiviral agents.76
Erythema multiforme appears to be a disease of the young, and the mildest form (erythema multiforme minor) characteristically features occurrence of erythematous papules on the skin that expand and enlarge peripherally, leaving a central vesicle or bulla and elevated circinate border (target lesions) (Figure 18). By definition, erythema multiforme minor may involve no more than one mucous membrane tissue, most often the oral mucosa (Figure 19). Often, lesions occur in the oral cavity with or without skin manifestations.77,78
Erythema multiforme major involves skin and two or more mucosal surfaces. Skin involvement may or may not be more extensive than in erythema multiforme minor.79
In a more severe form of erythema multiforme, Stevens-Johnson syndrome (SJS), skin involvement is extensive (as much as 10% of body area); the oral cavity, genitalia, and conjunctiva often are involved. SJS also may involve multiple organ systems, resulting in systemic symptoms. Often, the condition is precipitated by an adverse drug reaction and, occasionally, may be fatal (3% to 10%). SJS has been reported in the absence of skin lesions but with severe oral, ocular, genital, and systemic involvement.80
Toxic epidermal necrolysis (TEN), Lyell’s disease, is believed by some to represent the most severe form of the disease. It is characterized by high fever and widespread cutaneous vesiculobullous lesions involving more than 10% of bodily surfaces.82 It is usually initiated by an adverse drug reaction; however, mycoplasma pneumoniae infection also has been reported. Oral mucosal involvement can be severe, and loss of vision is a major risk. Fatalities range from 6.2% to 40%.81
Although a prodrome of flu-like symptoms has been described in erythema multiforme, onset of the diffuse inflammatory lesions is sudden and new lesions may continue to develop for days after onset. Without treatment, lesions tend to dissipate within 2 to 3 weeks; however, recurrent episodes may follow quickly, and some authors describe a chronic form of the disease.79
In the oral cavity, erythema multiforme characteristically features blister formation on the lips and oral mucosa, which rupture leaving severe erosions or ulcerations followed by formation of a grayish pseudomembrane, DG, and a hemorrhagic crust on the lips. Oral lesions may present with a parboiled or blanched appearance in perilesional areas. Nikolsky’s sign is usually positive.82
Histologic features of erythema multiforme are nonspecific. A monocytic t-lymphocyte perivascular submucosal infiltrate may be seen in early lesions, and inflammatory cells transmigrate the epithelium as keratinocyte degeneration occurs. Intraepithelial or subbasilar bulla separation may happen. Direct immunofluorescence is also nonspecific, revealing perivascular IgM and C3 along with granular deposits of IgG, C3, and fibrin in the basement membrane zone. Indirect immunofluorescence is negative. These features are suggestive of a cell-mediated immune response consistent with a hypersensitivity reaction.79,82,83
Erythema multiforme treatment usually consists of management of the triggering infection, discontinuation of drugs possibly responsible for the reaction, and systemic corticosteroids. Because primary herpetic gingivostomatitis may present with many features similar to erythema multiforme, the correct diagnosis must be established before use of corticosteroids. Consequently, early treatment may be palliative until the definitive diagnosis is determined.76
Surgical and Medical Management
The mucocutaneous disorders that most often affect the gingival mucosa are oral lichen planus, MMP, and pemphigus vulgaris. Linear IgA disease is an entity very similar in nature to MMP and also can affect the gingiva. Commonalities among these vesiculoerosive conditions are their inflammatory-mediated pathogeneses and probable autoimmune etiologies.
After the diagnosis of the vesiculoerosive disease is established, there are medication regimens that can be prescribed, which have been shown to alleviate the signs and symptoms by modulating the inflammatory pathways in disease pathogenesis. However, specific treatment modalities directed at correcting the underlying etiologies of these diseases have not been discovered.
The mainstay of treatment for all vesiculoerosive diseases of the oral cavity, and the most common first step in their management, is the use of corticosteroids, either taken systemically, injected perilesionally, or applied topically.84 Because of the potential for side effects associated with both topical and systemic long-term steroidal use, the treatment goal should be to achieve disease control with short, intermittent courses of therapy and maintenance with the lowest effective dose. Patients should be monitored after the withdrawal of therapy for the re-emergence of the signs and symptoms of the vesiculoerosive disease, and steroid treatment should be reinitiated as indicated. In addition, the practitioner should reinforce proper oral hygiene measures with the patient and institute frequent recalls for professional prophylaxis. This is because reduction in the overall inflammatory condition of the mucosa, including ordinary plaque-associated inflammation, may be beneficial for controlling this group of immune diseases.85,86
Topical gel preparations are most appropriate for oral mucosal use and can be mixed with Orabase® (Colgate-Palmolive, www.colgate.com) to increase their substantivity in the oral cavity, and include 0.1% triamcinolone, 0.05% fluocinonide, and 0.05% clobetasol.84 When the vesiculoerosive disease solely affects the gingiva and palate, it may be beneficial to use flexible custom delivery trays as an occlusive dressing, although this method may facilitate absorption through the ulcerated mucosa and thereby increase the risk of corticosteroid-associated systemic side effects. Dexamethasone elixir (0.5 mg/5 mL) is used as an oral rinse three to four times per day and can be swallowed or expectorated, depending on the therapeutic regimen used. Injectable dexamethasone may be delivered perilesionally for the treatment of discrete oral lesions; however, its applicability to the management of generalized oral vesiculoerosive disease manifested in the gingival tissues is obviously limited.84 Topical oral steroidal therapy increases the risk for candidal infection, and therefore, patients receiving topical steroidal treatment should be monitored and treated with antifungal medication appropriately.86,87
Vesiculoerosive disease that is refractory to local measures commonly is treated with systemic steroids.84 While no controlled studies support this option, there is nevertheless significant expert consensus validating this treatment. It is, in fact, considered the initial treatment of choice for PV.88 Burst therapy, consisting of high-dose, short-term use of prednisone with or without a taper, has been described as successful to achieve initial control over vesiculoerosive disease with minimal risk of side effects.89 A typical regimen would consist of 40 mg to 50 mg prednisone per day for 5 to 7 days. However, because it is possible that some side effects can be experienced with even a short-term course of therapy, specifically psychogenic and blood glucose control issues, case selection is paramount and should account for the medical profile of the patient.89
While corticosteroids can help control most cases of vesiculoerosive disease, trials of alternative therapeutic modalities may be desirable either when the practitioner wants to reduce the risk of steroid use (eg, steroid-sparing therapy) or when the patient’s disease is refractory to corticosteroids in appropriate doses.90 Medications, such as azathioprine, Dapsone, and mycophenolate, have been described widely in the literature as adjuvants to reduce the dosage of corticosteroids required to control the patient’s disease. However, the side-effect profiles are significant and their use is probably beyond the scope of most dental practitioners.84,89,90 Close comanagement with a physician qualified to monitor the patient and manage the side effects is advised. Tacrolimus, retinoids, and cyclosporine have a place in the management of vesiculoerosive disease; however, caution is advised to the practitioner unfamiliar with the use of these medications to treat immunologically mediated disease.87,88,90
Of concern to the dental practitioner who manages patients with these diseases is the friability of the gingiva and the concomitant degradation in proper oral hygiene measures. As noted previously, the resultant inflammation from plaque and calculus accumulation may not only enhance the progression of the underlying vesiculoerosive disease but also may contribute to the development of classic gingivitis and periodontitis, treatment of which is then in turn complicated by the gingival friability. It is a common observation that patients with these vesiculoerosive diseases have this self-perpetuating cycle of inflammation begetting inflammation.87-90
Though not examined in the literature, it has been the authors’ experiences that increasing medication dosage and/or frequency and tightening mucocutaneous disease control just before professional hygiene and surgical periodontal intervention may facilitate intraoperative tissue manipulation and reduce the occurrence of untoward surgical complications. Anecdotally, it may be beneficial to treat vesiculoerosive disease with a short course of topical steroids for several days before planned manipulation of the gingiva (periodontal surgery, scaling and root planing) to reduce the local inflammatory milieu that mediates the gingival friability that is typical in these patients. This hypothesis remains to be tested in an evidence-based format but offers fascinating research opportunities.
Conclusion
Patients presenting with mucocutaneous disorders can present challenges to the clinician, from medical management to surgical decision making. However, with advances in medical technology and research, patients with such disorders can have successful treatment, and morbidity associated with these conditions can be significantly reduced. Further research needs to be conducted in this area to minimize untoward effects from various treatment protocols and from the diseases themselves.
Disclosure
The opinions and assertions contained in this article are the private ones of the authors and are not to be construed as official or reflecting the views of the Department of the Navy, Department of Defense, or the US Government.
References
1. Position paper of the American Academy of Periodontology. Oral features of mucocutaneous disorders. J Periodontol. 2003;74(10):1545-1556.
2. Lo Russo L, Fedele S, Guiglia R, et al. Diagnostic pathways and clinical significance of desquamative gingivitis. J Periodontol. 2008;79(1):4-24.
3. Chan L. Oral manifestations of autoimmune blistering diseases. August 2006. eMedicine Web site. Available at: http://www.emedicine.com/derm/topic661.htm. Accessed May 28, 2008.
4. Al Hashimi I, Schifter M, Lockhart PB, et al. OLP and oral lichenoid lesions: diagnostic and therapeutic considerations. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103(suppl 1):S25-S31.
5. Chainani-Wu N, Silverman S, Lozada-Nur F, et al. OLP. Patient profile, disease progression and treatment responses. J Am Dent Assoc. 2001;132(7):901-909.
6. Rees TD. Unpublished research. Dallas, TX: Stomatology Center at Baylor College of Dentistry; 2007.
7. Eisen D. The clinical manifestations and treatment of OLP. Dermatol Clin. 2003;21(1):79-89.
8. Rogers RS 3rd, Eisen D. Erosive OLP with genital lesions. The vulvovaginal-gingival syndrome and the peno-gingival syndrome. Dermatol Clin. 2003;21(1):91-98.
9. Belfiore P, Di Fede O, Cabibi D, et al. Prevalence of vulval lichen planus in a cohort of women with OLP: an interdisciplinary study. Br J Dermatol. 2006;155(5):994-998.
10. Petruzzi M, De Benedittis M, Pastore L, et al. Peno-gingival lichen planus [Published eratum in: J Periodontol. 2006;77(2):327]. J Periodontol. 2005;76(12):2293-2298.
11. Andreason JO. OLP: a clinical evaluation of 115 cases. Oral Surg Oral Med Oral Pathol. 1968;25(1):31-41.
12. Edwards PC, Kelsch R. OLP: clinical presentation and management. J Can Dent Assoc. 2002;68(8):494-499.
13. van der Meij, Mast H, van der Wall I. The possible premalignant character of OLP and oral lichenoid lesions: a prospective five year follow-up study of 192 patients. Oral Oncol. 2007;43(8):742-748.
14. Soto Araya M, Rojas Alcayaga G, Esquip A. Association between psychological disorders and presence of OLP, burning mouth syndrome, and recurrent aphthous stomatitis. Med Oral. 2004;9(1):1-7.
15. Koray M, Dülger O, Ak G, et al. The evaluation of anxiety and salivary cortisol levels in patients with OLP. Oral Dis. 2003;9(6):298-301.
16. Chaudhary S. Psychosocial stressors in OLP. Aust Dent J. 2004;49(4):192-195.
17. Lodi G, Scully C, Carrozzo M, et al. Current controversies in OLP: report of an international consensus meeting. Part 1. Viral infections and etiopathogenesis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100(1):40-51.
18. Lamey P-J, Gibson J, Barclay SC, et al. Grinspan’s syndrome: a drug induced phenomenon? Oral Surg Oral Med Oral Pathol. 1990;70(2):184-185.
19. Kalmar J. Oral manifestations of drug reactions. October 2006. eMedicine Web site. Available at: http//www.emedicine.com/DERM/topic816.htm. Accessed May 28, 2008.
20. Torgerson RR, Davis MDP, Bruce AJ, et al. Contact allergy in oral disease. J Am Acad Dermatol. 2007;57(2):315-321.
21. Fowler CB, Rees TD, Smith BR. Squamous cell carcinoma on the dorsum of the tongue arising in a long-standing lesion of erosive lichen planus. J Am Dent Assoc. 1987;115(5):707-710.
22. Schubert MM, Correa ME. Oral graft-versus-host disease. Dent Clin North Am. 2008;52(1):79-109.
23. Treister NS, Cook EF Jr, Antin J, et al. Clinical evaluation of oral chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2008;14(1):110-115.
24. Imanguli MM, Pavietic SZ, Guadagnini JP, et al. Chronic graft versus host disease of oral mucosa. Review of available therapies. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101(2):175-183.
25. Byun JH, Park BW, Kim JR, et al. Squamous cell carcinoma of the tongue after bone marrow transplant and graft-versus-host disease: a case report and review of the literature. J Oral Maxillofac Surg. 2008;66(1):144-147.
26. Islam MN, Cohen DM, Ojha J, et al. Chronic ulcerative stomatitis: diagnostic and management challenges—four new cases and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;104(2):194-203.
27. Lorenzana ER, Rees TD, Glass M, et al. Chronic ulcerative stomatitis: a case report. J Periodontol. 2000;71(1):104-111.
28. Solomon LW, Neiders ME, Zwick MG, et al. Autoimmunity to deltaNp63alpha in chronic ulcerative stomatitis. J Dent Res. 2007;86(9):826-831.
29. Rinaggio J, Crossland DM, Aeid MY. A determination of the range of oral conditions submitted for microscopic and direct immunofluorescence analysis. J Periodontol. 2007;78(10):1904-1910.
30. Beutner EH, Chorzelski TP, Parodi A, et al. Ten cases of chronic ulcerative stomatitis with stratified epithelium-specific antinuclear antibody. J Am Acad Dermatol. 1991;24(5 pt 1):781-782.
31. Parodi A. Cozzani E, Massone C, et al. Prevalence of stratified epithelium-specific antibodies in 138 patients with lichen planus. J Am Acad Dermatol. 2007;56(6):974-978.
32. Chorzelski TP, Olszewska M, Jarzabek-Chorzelska M, et al. Is chronic ulcerative stomatitis an entity? Clinical and immunological findings in 18 cases. Eur J Dermatol. 1998;8:261-265.
33. Scully C, Lo Muzio L. Oral mucosal diseases: mucous membrane pemphigoid. Br J Oral Maxillofac Surg. 2007 Sep 3 [epub ahead of print].
34. Rashid KA, Gürcan HM, Ahmed AR. Antigen specificity in subsets of mucous membrane pemphigoid. J Invest Dermatol. 2006:126(12):2631-2636.
35. Ojha J, Bhattacharyya I, Stewart C, et al. Cicatricial pemphigoid with severe gingival and laryngeal involvement in an 18-year-old female. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;104(3):363-367.
36. Cheng YS, Rees TD, Wright JM, et al. Childhood oral pemphigoid: a case report and review of the literature. J Oral Pathol Med. 2001;30(6):372-377.
37. Lourenço SV, Boggio P, Agner Macado Martins LE, et al. Childhood oral mucous membrane pemphigoid presenting as desquamative gingivitis in a 4-year-old girl. Acta Derm Venereol. 2006;86(4):351-354.
38. Endo H, Rees TD, Kuyama K, et al. Clinical and diagnostic features of mucous membrane pemphigoid. Compend Contin Educ Dent. 2006;27(9):512-518.
39. Suresh L, Kumar V. Significance of IgG4 in the diagnosis of mucous membrane pemphigoid. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;104(3):359-362.
40. Torchia D, Caprioni M, Fabbri P. Linear IgA dermatosis with exclusive mucosal involvement or mucous membrane pemphigoid? Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;104(2):151-152.
41. Solomon LW, Yerke LM, Kumar V. Differentiation of mucous membrane pemphigoid subgroups with confocal imaging. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;104(6):790-795.
42. Calabresi V, Carrozzo M, Cozzani E, et al. Oral pemphigoid autoantibodies preferentially target BP 180 ectodomain. Clin Immunol. 2007;122(2):207-213.
43. Sadler E, Lazarova Z, Sarasombath P, et al. A widening perspective regarding the relationship between anti-epiligrin cicatricial pemphigoid and cancer. J Dermatol Sci. 2007;47(1):1-7.
44. Lewis MA, Yaqoob NA, Emanuel C, et al. Successful treatment of oral linear IgA disease using mycophenolate. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103(4):483-486.
45. Porter SR, Bain SE, Scully C. Linear IgA disease manifesting as desquamative gingivitis. Oral Surg Oral Med Oral Pathol. 1992;74(2):179-182.
46. Femiano F, Scully C, Gombos F. Linear IgA dermatosis induced by a new angiotensin-converting enzyme inhibitor. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;95(2):169-173.
47. Cauza K, Hinterhuber G, Sterniczky B, et al. Unusual clinical manifestations of linear IgA dermatosis: a report of two cases. J Am Acad Dermatol. 2004;51(2 suppl):S112-S117.
48. Eguia del Valle A, Aguirre Urízar JM, Martínez Sahuquillo A. Oral manifestations caused by the linear IgA disease. Med Oral. 2004;9(1):39-44.
49. Sánchez AR, Rogers RS 3rd, Kupp LI, et al. Desquamative gingivitis associated with IgG/IgA pemphigoid presents a challenging diagnosis and treatment: a case report. J Periodontol. 2004;75(12):1714-1719.
50. Nanda A, Dvorak R, Al-Sabah H, et al. Linear bullous disease of childhood: an experience from Kuwait. Pediatr Dermatol. 2006;23(5):443-447.
51. Usmani N, Baxter KF, Child JA, et al. Linear IgA disease in association with chronic lymphocytic leukaemia. Br J Dermatol. 2004;151(3):710-711.
52. Nanda A, Dvorak R, Al-Sabah H, et al. Association of linear IgA bullous disease of childhood with Crohn’s disease. Int J Dermatol. 2006;45(10):1184-1186.
53. Schultewolter T, Goos M, Dissemond J. Linear IgA dermatosis in an immunosuppressed patient after allogenic bone marrow transplantation. J Eur Acad Dermatol Venereol. 2004;18(6):721-725.
54. Diaz LA, Prisayanh, PS, Dasher DA, et al. The IgM anti-desmoglein 1 response distinguishes Brazilian pemphigus foliaceus (fogo selvagem) from other forms of pemphigus. J Invest Dermatol. 2008;128(3):667-675.
55. Yokokura H, Demitsu T, Kakurai M, et al. Paraneoplastic pemphigus mimicking erosive mucosal lichen planus associated with primary hepatocellular carcinoma. J Dermatol. 2006;33(12):842-845.
56. Zhu X, Zhang B. Paraneoplastic pemphigus. J Dermatol. 2007;34(8):503-511.
57. Scully C, Challacombe SJ. Pemphigus vulgaris: update on etiopathogenesis, oral manifestations, and management. Crit Rev Oral Biol Med. 2002;13(5):397-408.
58. Mahajan VK, Sharma NL, Sharma RC, et al. Twelve-year clinico-therapeutic experience in pemphigus: a retrospective study of 54 cases. Int J Dermatol. 2005;44(10):821-827.
59. Lamey PJ, Rees TD, Binnie WH, et al. Oral presentation of pemphigus vulgaris and its response to systemic steroid therapy. Oral Surg Oral Med Oral Pathol. 1992;74(1):54-57.
60. Mignogna MD, Lo Muzio L, Bucci E. Clinical features of gingival pemphigus vulgaris. J Clin Periodontol. 2001;28(5):489-493.
61. Endo H, Rees TD, Matsue M, et al. Early detection and successful management of oral pemphigus vulgaris: a case report. J Periodontol. 2005;76(1):154-160.
62. Presland R, Dale BA. Epithelial structural proteins on the skin and oral cavity: function in health and disease. Crit Rev Oral Biol Med. 2000;11:383-408.
63. Endo H, Rees TD, Hallmon WW, et al. Disease progression from mucosal to mucocutaneous involvement in a patient with desquamative gingivitis associated with pemphigus vulgaris. J Periodontol. 2008;79(2):369-375.
64. Scully C, Mignogna M. Oral mucosal disease: pemphigus. Br J Oral Maxillofac Surg. 2008;46(4):272-277.
65. Browne C. Lupus erythematosus, acute. November 2006. eMedicine Web site. Available at: http://www.emedicine.com/DERM/topic245.htm. Accessed May 28, 2008.
66. Lourenço SV, Nacaqami Sotto M, Constantino Vilela MA, et al. Lupus erythematosus: clinical and histopathological study of oral manifestations and immunohistochemical profile of epithelial maturation. J Cutan Pathol. 2006;33(10):657-662.
67. Jayakumar ND, Jaiganesh R, Padmalatha O, et al. Systemic lupus erythematosus. Indian J Dent Res. 2006;17(2):91-93.
68. Messadi DV, Waibel JS, Mirowski GW. White lesions of the oral cavity. Dermatol Clin. 2003;21(1):63-78.
69. Werth VP. Clinical manifestations of cutaneous lupus erythematosus. Autoimmun Rev. 2005;4(5):296-302.
70. Rees TD. Adjunctive therapy. In: Nevins M, Becker W, Kornman K, eds. Proceedings of the World Workshop in Clinical Periodontics. Chicago, IL: American Academy of Periodontology; 1989:X1-X31.
71. McCarty DJ, Manzi S, Medsger TA Jr, et al. Incidence of systemic lupus erythematosus. Race and gender differences. Arthritis Rheum. 1995;38(9):1260-1270.
72. López-Labady J, Villarroel-Dorrego M, González N, et al. Oral manifestation of systemic and cutaneous lupus erythematosus in a Venezuelan population. J Oral Pathol Med. 2007;36(9):524-527.
73. Orteu CH, Buchanan JAG, Hutchison I, et al. Systemic lupus erythematosus presenting with oral mucosal lesions: easily missed? Br J Dermatol. 2001;144(6):1219-1223.
74. Ruokenen H, Malström M, Studd S. Factors influencing the recurrence of erythema multiforme. Proc Finn Dent Soc. 1988;84(3):167-174.
75. Lozada F, Silverman S Jr. Erythema multiforme. Clinical characteristics and natural history in fifty patients. Oral Surg Oral Med Oral Pathol. 1978;46(5):628-636.
76. Woo SB, Challacombe SJ. Management of recurrent oral herpes simplex infections. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103(suppl 1):S12-S18.
77. Jawetz R, Eikin Avigayll E, Michael L, et al. Erythema multiforme limited to the oral mucosa in a teenager on oral contraceptive therapy. J Pediatr Adolesc Gynecol. 2007;20(5):309-313.
78. Rees TD. Phenothiazine—another possible etiologic agent in erythema multiforme. Report of a case. J Periodontol. 1985;56(8):480-483.
79. Farthing P, Bagan JV, Scully C. Mucosal diseases series. Number IV. Erythema multiforme. Oral Dis. 2005;11(5):261-267.
80. Vanfleteren I, Van Gysel D, De Brandt C. Stevens-Johnson syndrome: a diagnostic challenge in the absence of skin lesions. Pediatr Dermatol. 2003;20(1):52-56.
81. Yamane Y, Aihara M, Ikezawa Z. Analysis of Stevens-Johnson syndrome and toxic epidermal necrolysis in Japan from 2000 to 2006. Allergol Int. 2007;56(4):419-425.
82. Scully C, Bagan J. Oral mucosal diseases: erythema multiforme. Br J Oral Maxillofac Surg. 2008;46(2):90-95.
83. Ayangco L, Rogers RS 3rd. Oral manifestations of erythema multiforme. Dermatol Clin. 2003;21(1):195-205.
84. Siegel MA, Silverman S, Sollecito TP, eds. American Academy of Oral Medicine’s Clinician’s Guide to Treatment of Common Oral Conditions. 5th ed. Lewiston, NY: BC Decker, Inc; 2001.
85. Sollecito TP, Parisi, E. Mucous membrane pemphigoid. Dent Clin North Am. 2005;49(1):91-106.
86. Eisen D. The clinical manifestations and treatment of OLP. Dermatol Clin. 2003;21(1):79-89.
87. N, Silverman S Jr, Lozada-Nur F, et al. OLP: patient profile, disease progression and treatment responses. J Am Dent Assoc. 2001;132(7):901-909.
88. Stanley JR. Therapy of pemphigus vulgaris. Arch Dermatol. 1999;135(1):76-78.
89. Silverman S Jr, Lozada-Nur F, Migliorati C. Clinical efficacy of prednisone in the treatment of patients with oral inflammatory ulcerative diseases: a study of fifty-five patients. Oral Surg Oral Med Oral Pathol. 1985;59(4):360-363.
90. Fine JD. Management of acquired bullous skin diseases. N Eng J Med. 1995;333(22):1475-1484.